CADBURE is a bioinformatics tool for evaluating spliced aligner performance on the user data. It compares the given two alignment results based on the relative reliable mapping and advantageous mapping in the predefined scenarios. It outputs the read name lists associated with the scenarios, so the user can view the mappings in any BAM viewer.
Why should I use CADBURE?In RNA-Seq study for differential expression, mapping of cDNA data set to the reference genome or transcriptome is an important step that will decide the outcome of your study. There are plenty of spliced aligners available ( http://wwwdev.ebi.ac.uk/fg/hts_mappers/) and choosing one that will give better performance on your data is not trivial. Moreover, aligner's performance depends on the properties of data. The solution is evaluating spliced aligner performance on your data to decide on one. CADBURE is an easy to use and simple tool for comparing alignment results. CADBURE outputs discrete measures of specificity and accuracy for each compared aligner which will help you decide. Moreover, CADBURE distribution comes with R script that will help you do bootstrap stats on the difference of measures (both specificity and accuracy) to delineate statistical significance.
How can I cite CADBURE?
If you use CADBURE please cite,
Praveen Kumar Raj Kumar, Thanh V. Hoang, Michael L. Robinson, Panagiotis A. Tsonis, Chun Liang: CADBURE: A generic tool to evaluate the performance of spliced aligners on RNA-Seq data, in press. Scientific Reports, 5:13443, DOI: 10.1038/srep13443.
CADBURE itself does not need installation as long as Perl is installed. However you would need two Perl Modules (Bio::DB::Sam and HTML::Table), available from CPAN (http://www.cpan.org/).
How do I execute CADBURE?
You can execute it like
./CADBURE
Or
perl CADBURE
Executing it without the flags prints the usage.
Alignment results should be in BAM format. For faster processing we recommend the input to be sorted based on read. See samtools ( http://samtools.sourceforge.net/) for how to do the conversion of SAM format to BAM format.
Is there any limit for size of BAM file?No. There is no limit.
I see a huge number for particular scenario in an aligner. How can I see those mappings?
Find the corresponding “scenario_number” folder and inside it you can see files
containing list of reads involved in the scenario for your aligner. Depending upon
the scenario there can be either one file or two files for the aligners. In either
case the file name should say this. Upon opening your BAM file of interest in any
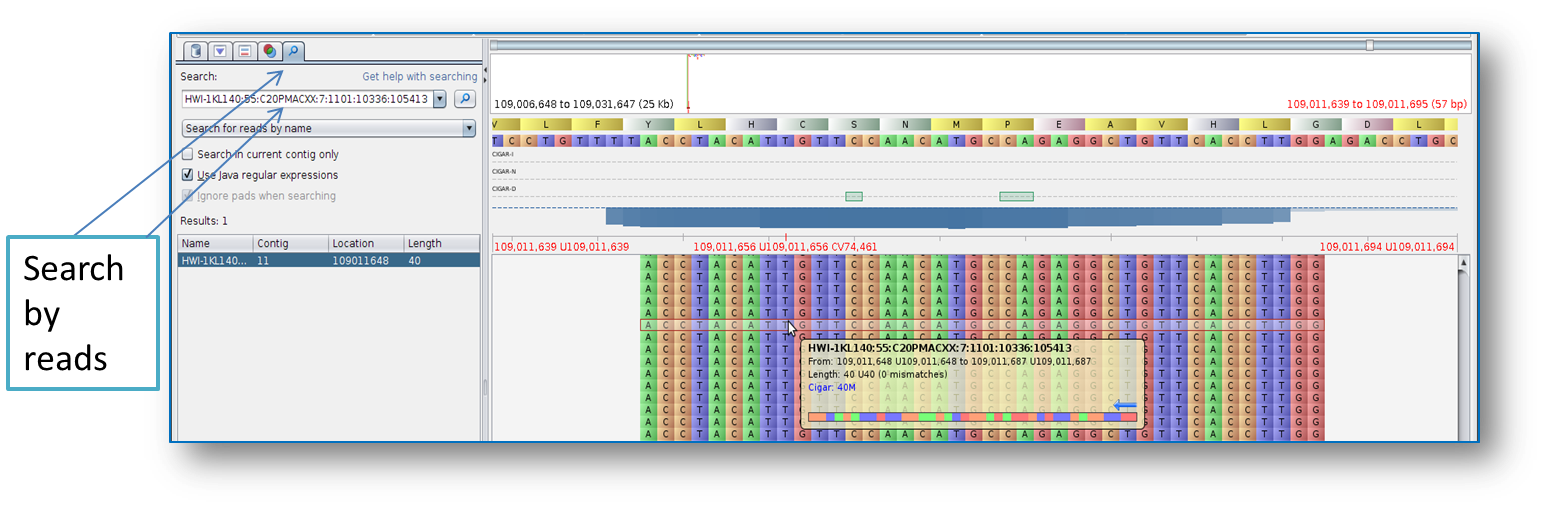
BAM viewer like Tablet
(
http://ics.hutton.ac.uk/tablet/) for example, use the read to search for
mapping (Figure 1).
This probably means your system has old version of Genome modelling tools called Pindel (http://gmt.genome.wustl.edu/packages/pindel/). Easy fix for this problem is that to get Bio-SamTools from here https://github.com/gitpan/Bio-SamTools.
I get an error like "Unable to create Scenario_1"?Please make sure you have proper permissions to create folders. If not use “sudo” to execute CADBURE.
Whom to contact to report a probelm?Please contact the developer, Praveen [praveenk at pitt.edu] OR the lab direcor, Chun Liang [liangc at miamioh.edu]